Our latest work has now been published in Journal of Cell Science:
A general role for TANGO1, encoded by MIA3, in secretory pathway organization and function
What’s more for the first time in ages, we have a cover. OK, the online cover. Still counts though. I won’t restate the paper here but instead will reflect on my favourite parts and why we don’t think our data support models where TANGO1 is selective for procollagen. Please do read the full paper for the background, detail, and greater context.
I have worked on the system governing the export of proteins from the endoplasmic reticulum in mammalian cells since 1999 and with the team in Bristol since 2001. While I think our lab has made some significant contributions, we have of course always followed the field with great interest. A part we have always been reluctant to tackle from has been TANGO1, encoded by Mia3. Initially reported as a co-factor for procollagen secretion in 2009, substantial data since then has elaborated this role to include some other large cargoes, more nuanced mechanisms involving engagement of Hsp47 (a key chaperone) rather than procollagen itself, and a broader role at the point of ER exit through recruitment of a tethering complex located at the ER-Golgi Intermediate compartment (ERGIC) the first post-ER compartment. You can read up on work to date regarding the ER-Golgi interface and work to date in our review article in Trends in Cell Biology.
With the advent of CRISPR-Cas9 gene editing and Janine McCaughey’s inexhaustible thirst for new data, we embarked on a project to edit the genome of human cells to try and eliminate expression from the Mia3 gene that includes TANGO1. The paper goes into much more depth here of course but the key issue is that there are two major (and likely several minor) isoforms of TANGO1, the shorter of which has only a small region within the lumen of the ER. This has previously been shown (by RNAi and other work) to be sufficient to support secretion of procollagen.
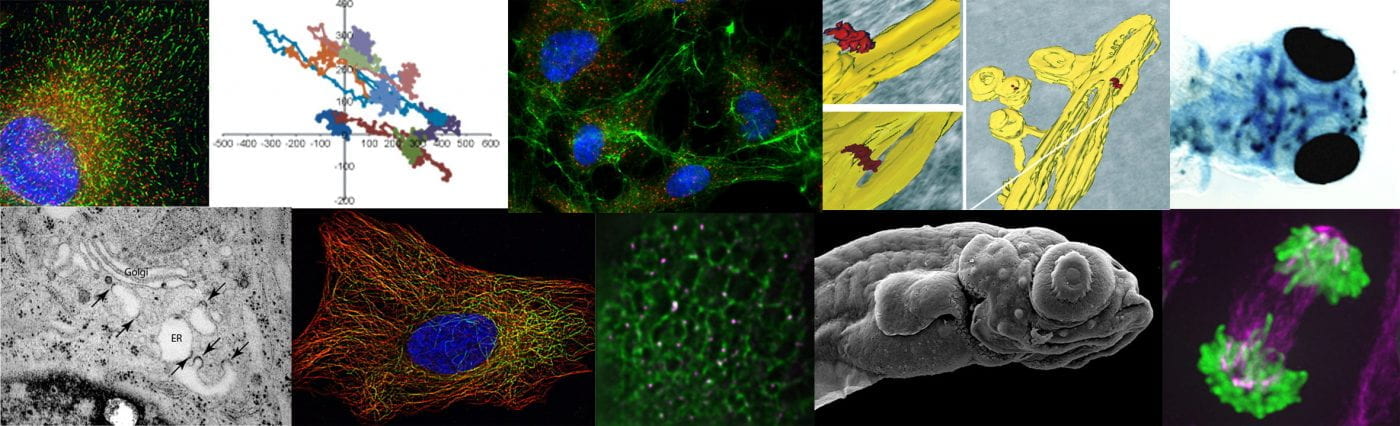
The most effective “knockout” that we have almost (but not quite) eliminates expression fo all isoforms. This showed a dramatic phenotype (which made the cover) of a multitude of vesicular structures at the interface of the ER and Golgi.

I love this image as it is so thought-provoking and generates ideas every time I look at it. My own speculation here (I use that word advisedly based on feedback!) is that TANGO1 constrains the formation of COPII-coated vesicles to generate a functional ERGIC. In its absence, COPII vesicle formation is more efficient (maybe like in other organisms that don’t express TANGO1 or really have a definable ERGIC????) and that leads to this phenotype. Trafficking works perfectly well in organisms that don’t have TANGO1 but they maybe don’t have the same secretory load as vertebrates. So, I see TANGO1 as an “optimiser” of the system, not as a de facto procollagen/large cargo packaging factor.
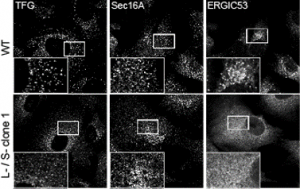
In my mind I see this most closely aligned to the data in the paper that shows a “collapse” of the ERGIC with markers ERGIC53 and SURF4 becoming exclusively localized to the ER. my interpretation – we have lost the ability to either build or maintain a functional ERGIC. Maybe these vesicles represent a fragmented Golgi? Maybe (and we’re trying some immuno-EM of course) but this phenotype doesn’t look like a COPI block or other Golgi-based perturbations that I have seen.
Why does this matter? Many organisms get along fine without a definable ERGIC (but then many of these same organisms get along fine with a very differently organization of the Golgi). The previous data on the role of TANGO1 in recruiting the ERGIC becomes key here. What if in fact its role is to support biogenesis of the ERGIC? What if the role of the ERGIC is to support efficient cargo secretion? Any loss of integrity here would lead to defects in secretion which we see in multiple assays in the paper, including proteomics.
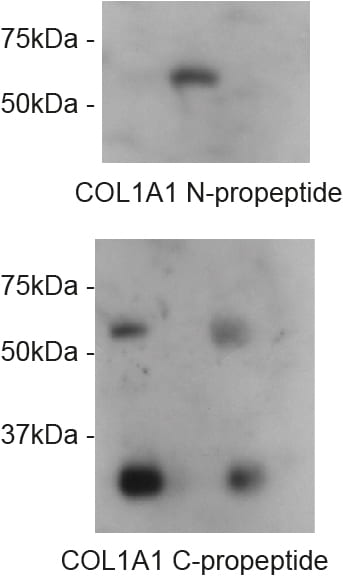
The impact on secretion here is significant. Surely if we block ER export then this is what you would expect to see? Well, ER export isn’t blocked. Indeed, our proteomics data show enhanced secretion of some cargoes. For the majority of “canonical” cargoes secretion is slowed. This is also true for procollagen which is seen to accumulate inside cells (the “L” lanes on the blot below).
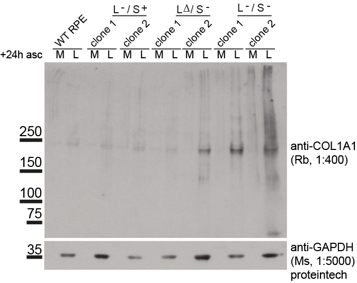
Loss of TANGO1 causes procollagen to accumulate inside cells. The severity of the defect correlates with the severity of disruption to the Mia3 locus. So where does this leave us in terms of our interpretation that TANGO1 is NOT procollagen- or “large cargo”-selective?
My favoured model here is that procollagen is one of the most significantly impacted cargoes – defects in its secretion are seen on perturbation of almost any key component of the early secretory pathway from COPII subunits (e.g. Sec13 and Sec23), components of ER-Golgi tethers e.g. Sedlin and NBAS, and Golgi proteins such as GMAP210, giantin, and GORAB.
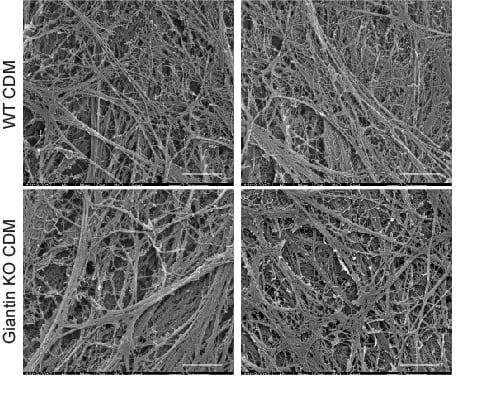
In our data we show that many other cargoes are affected, small soluble proteins, larger matrix proteins, and transmembrane proteins. We’ve used the RUSH system to retain and then control release of cargo (including procollagen) from the ER – this leads to the critique that we are overexpressing and that we are forcing a large synchronous cargo load. Fair enough but we have back up these data with other methods – the blot here is on endogenous procollagen, proteomics of the soluble “secretome” from these cells as well as of the cell-derived matrix show major changes in secretion.
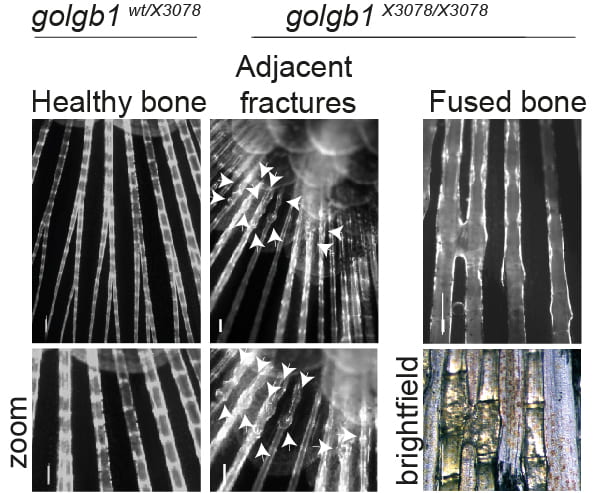
Another key factor we find with regard to reduced secretion of collagen in general is that the expression of many collagens is downregulated. The RNAseq data are a key component of this work and one that we have yet to explore fully. Type XI collagen is strongly affected here and is critical for the formation of a fibrillar matrix. This could impact key defects seen with in vivo mouse models and in humans (here and here) with disrupted Mia3 expression. We do not underestimate the likely impact of changes in gene expression to the phenotypes we see.
Importantly we can also restore key phenotypes with the short form of TANGO1 which suggests that engagement with Hsp47/procollagen might not be required for at least some of this.
I reflect back to our reviews on this particularly the one in Trends in Cell Biology where we discuss the importance of the ERGIC as a “buffer” to maintain compartment identity of the ER and the Golgi. I think there is also now more of a consensus that a more amorphous membrane network traffics cargo through the early secretory pathway and our contention is that TANGO1 is a key component that sets up this membrane network. Much of this also echoes back to work, notably from Hans-Peter Hauri (now retired) on the identity of the ERGIC. Our data indicate that it is not a stable compartment but requires TANGO1 for its existence – whether this is really in terms of ERGIC biogenesis or maintenance remains to be defined.
This of course is not the whole story by any means. We are now developing this work thanks to further UKRI-BBSRC funding and will be able to explore phenotypes in vivo thanks to a new collaboration with Brian Link at the Medical College of Wisconsin who has made several zebrafish models of relevance here. We hope that the proteomics and genomics data in this paper will inform much of this work. We also of course make that data freely available to all, such that anyone can build on this study. Plasmids are also in Addgene for those interested.
I hope you will read the full paper. As usual – a great experience publishing with Journal of Cell Science (COI – I am an Editor but it really is a great experience!).