Our new preprint is now available on bioRxiv. We are really excited to share this new work from Nicola Stevenson which comes from our great collaboration with Chrissy Hammond (@ChrissyLHam) and Dylan Bergen (@SciBergen) https://www.biorxiv.org/content/10.1101/2020.05.25.115279v1
Some background:
Giantin is a golgin, it is ubiquitously expressed and conserved throughout vertebrates at least. Golgins define the specificity of incoming vesicle trafficking to the Golgi (beautiful work from Mie Wong and Sean Munro (Wong and Munro, 2014 https://science.sciencemag.org/content/346/6209/1256898/tab-article-info).
However, in their work, they have not been able to assign a role for giantin as a vesicle tether. Our own work has shown that loss of giantin results in minor defects in Golgi organization but is required for normal gene expression of some glycosyltransferases, notably GALNT3. doi: 10.1242/jcs.212308
It is also required for normal cilia formation in cells (Asante et a., 2013; Stevenson et al., 2017, Bergen et al., 2017)
(https://jcs.biologists.org/content/126/22/5189 and https://jcs.biologists.org/content/131/9/jcs212258) and zebrafish (https://bio.biologists.org/content/6/8/1180).
Various model organisms with mutations in or deletions of giantin, including our zebrafish models, show consistent defects in extracellular matrix formation. We sought to define this in more detail using our own zebrafish mutants and giantin KO cell lines.
The new data:
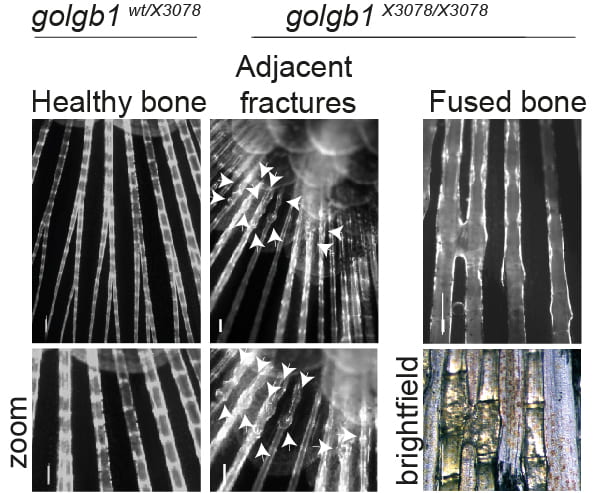
We find that giantin mutant zebrafish are prone to spontaneous fractures and bone mineralization defects. There is also a small but reproducible reduction in expression of type I procollagen in these tissues.
To look at this in more detail we switched to our giantin KO cells where we had also identified a consistent decrease in COL1A1 gene expression. Despite that, to our surprise we found a clear INCREASE in type I procollagen protein in these cells.
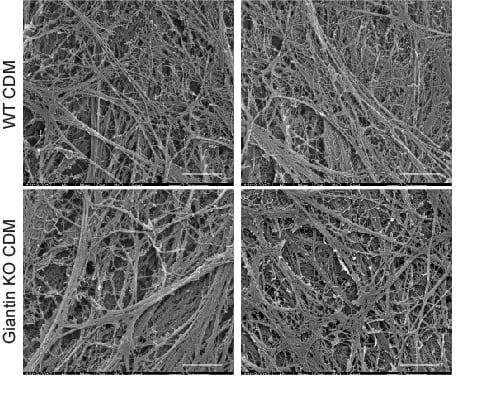
This was accompanied by increased secretion of type I procollagen and small but clear defects in the organization of extracellular matrix derived from these cells.
Using live cell imaging, we were surprised to find that these cells showed no apparent differences in trafficking of pro-α1(I) compared to controls.
Now we come to the most exciting part for me – what we did find was a clear defect in N-terminal processing of the N-terminal propeptide of pro-α1(I). Specifically, that this part of the precursor is NOT effectively cleaved in giantin KO cells.
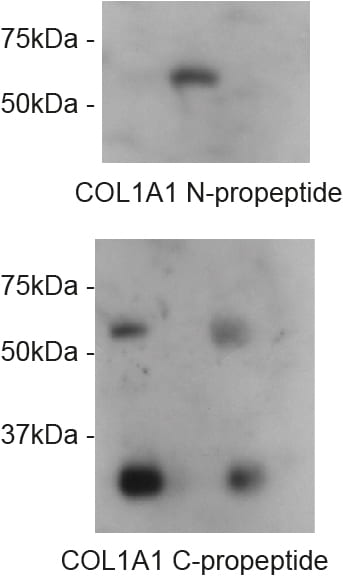
N-pro is retained until pro-α1(I) reaches the extracellular space. Most significantly, the N-propeptide is readily detected in cell lysates of WT cells but not in giantin KO cells. Our GFP-tagged collagen that we have used to define the intracellular trafficking route reports this really effectively using a variety of tags.
No defects were seen in processing of the C-terminal propeptide.
This finding was robustly reproduced in Nikki’s experiments and indeed, because of possible issues around clonal selection of our KO cells and the fact we only had one line, Nikki then generated further giantin KO lines.

Really intriguingly, these all show procollagen processing defects but each in a slightly different way. This supports our idea that this is not a direct mechanistic link between giantin and procollagen processing but rather reflects a fundamental defect at the Golgi.
We have spent years chasing the N-pro enzymes, especially the ADAMTS proteins but we haven’t found antibodies that detect them robustly nor have we been able to generate any tagged forms of ADAMTS2/3/14 that leave the ER.
There is also a lot of other data in here and mentions of what hasn’t worked or defined clear outcomes. Gaps and questions aplenty!
Can we define a direct link between giantin and the processing machinery? No. That said, we have to question whether our current understanding of ADAMTS biology is accurate (what if they are in fact primarily localized to the ER?).
We also can’t prove that the bone defects including susceptibility to fracture is directly linked to the N-pro defect. This is primarily because we can’t detect pro-α1(I) by immunoblotting in fish.
So what do we know from this?
- Giantin is required for normal bone integrity in zebrafish
- Giantin is also required for normal N-terminal propeptide cleavage of pro-α1(I)
Relationship to other work:
There is precedent for detection of intracellular procollagen processing in both chick and mouse tendons (Canty‐Laird et al., 2012; Canty et al., 2004; Humphries et al., 2008). Here, Kadler and colleagues found processed forms of type I procollagen in a detergent soluble fraction from tendon explants, consistent with an intracellular pool. This is however, also consistent with a detergent soluble extracellular pool.
In other experiments, his lab showed the presence of the N-propeptide in the presence of brefeldin A (which causes merging of the Golgi with the ER) (Canty-Laird et al., 2012). We consider that this shows clearly that the N-pro “can” be cleaved inside cells when you force the enzyme and substrate together using BFA.
We consider that our data shows that giantin is needed for this cleavage event to occur inside cells. This is the first defined “function” for giantin and clear support for the concept of intracellular processing of type I procollagen.
So why don’t we see a clearer phenotype in giantin mutant fish? Why doesn’t this reflect an EDS type VII phenotype (which arises from mutations in the N-pro enzyme)? The most likely explanation is that extracellular processing can occur but is inefficient.
If that is all true, then why would some pro-α1(I) be cleaved inside the cell if all can be cleaved outside? This raises the possibility of different pools of procollagen that are cleaved at different locations or at different times.
Could this relate to the ultimate role of that collagen? Maybe tissue formation versus repair? Could this relate to circadian control of matrix composition? We don’t know but we’re working on these and more ideas right now as part of our BBSRC-funded work with Karl and his colleagues in Manchester.
Is this work “perfect”? No. Is this complete? No. BUT we can say it is really robust, provides some key insight that will hopefully allow us and others to build on to better understand collagen matrix assembly and repair.
References:
Asante, D., L. Maccarthy‐Morrogh, A.K. Townley, M.A. Weiss, K. Katayama, K.J. Palmer, H. Suzuki, C.J.
620 Westlake, and D.J. Stephens. 2013. A role for the Golgi matrix protein giantin in ciliogenesis through
621 control of the localization of dynein‐2. J Cell Sci. 126:5189‐5197.
Canty‐Laird, E.G., Y. Lu, and K.E. Kadler. 2012. Stepwise proteolytic activation of type I procollagen to
648 collagen within the secretory pathway of tendon fibroblasts in situ. Biochem J. 441:707‐717.
Canty, E.G., Y. Lu, R.S. Meadows, M.K. Shaw, D.F. Holmes, and K.E. Kadler. 2004. Coalignment of plasma
652 membrane channels and protrusions (fibripositors) specifies the parallelism of tendon. J Cell Biol.
653 165:553‐563.
Humphries, S.M., Y. Lu, E.G. Canty, and K.E. Kadler. 2008. Active negative control of collagen fibrillogenesis
694 in vivo. Intracellular cleavage of the type I procollagen propeptides in tendon fibroblasts without
695 intracellular fibrils. J Biol Chem. 283:12129‐12135.
Stevenson, N.L., D.J.M. Bergen, R.E.H. Skinner, E. Kague, E. Martin‐Silverstone, K.A. Robson Brown, C.L.
779 Hammond, and D.J. Stephens. 2017. Giantin‐knockout models reveal a feedback loop between Golgi
780 function and glycosyltransferase expression. J Cell Sci. 130:4132‐4143.
Wong, M., and S. Munro. 2014. Membrane trafficking. The specificity of vesicle traffic to the Golgi is
793 encoded in the golgin coiled‐coil proteins. Science. 346:1256898.